Next generation sequencing (NGS) has become a powerful tool to identify genetic variants and variable gene expression patterns that correlate with disease state and provide clinically-relevant mechanistic insights. The entire NGS workflow can be broken down into four steps: sample extraction, library preparation, sequencing, and analysis
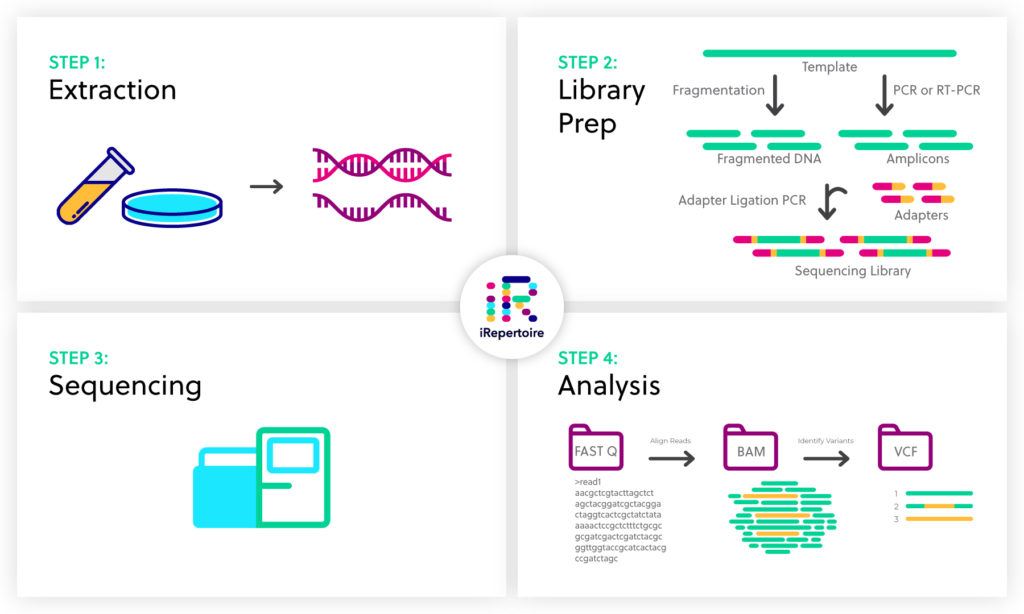
Step 1: Sample extraction
NGS can be performed on any sample that yields DNA or RNA (e.g., cell cultures, fresh-frozen tissues, formalin-fixed paraffin-embedded (FFPE) tissues, blood, saliva, and bone marrow). Various extraction protocols based on the starting material are available, and generally each extraction method has been optimized to yield the highest quality and largest amount of nucleic acid from the respective sample type. Following extraction, the amount and quality of DNA or RNA should be determined, as high-quality starting material is critical for successful sequencing. The purity of a nucleic acid sample is typically expressed as an A260/280 value, which can be determined using a spectrophotometer such as Thermo Scientific’s Nanodrop (Cat. No. ND-8000-GL). “Pure” DNA generally reports an A260/280 reading of 1.8, whereas RNA is closer to 2.0
To learn more about sample extraction for immune repertoire sequencing, see our sample preparation guide
Step 2: Library preparation
Preparation of a sequencing library from your RNA or DNA sample involves two basic steps: 1) amplification to yield a pool of appropriately sized target sequences, and 2) the addition of sequencing adapters that will later interact with the NGS platform. If RNA is the starting template, an additional step is needed in which the RNA is first converted to cDNA by reverse transcription.
PCR amplification yields a collection of specifically sized DNA fragments – called a library – that are compatible with the sequencing system to be used. The primers used in library preparation are designed based on the sequences of interest, which range from a whole genome to particular RNA transcripts.
The adapter ligation step essentially bookends the amplified DNA or cDNA fragments, called amplicons, with specific oligonucleotide sequences that will interact with the surface of a sequencing flow cell. If multiple samples are to be sequenced in a single sequencing run, a unique identifier, or barcode, is additionally ligated to the amplicon. The resulting completed libraries can be pooled into a single sequencing run that is then “demultiplexed” during data analysis.
For specific information about how to prepare a sequencing library for the immune repertoire, see our page on immune repertoire sequencing.
Step 3: DNA sequencing
Parallel sequencing is performed using an NGS platform. The library is loaded onto the sequencer which then “reads” the nucleotides one by one. The number of reads produced will vary depending on the sequencing platform and kit used. Several methods of NGS have been developed including pyrosequencing, sequencing by ligation (SOLiD), sequencing by synthesis (SBS – Illumina), and Ion Torrent sequencing. Illumina sequencing is by far the most popular, resulting in 90% of the world’s sequencing data (data per Illumina’s website).
Illumina Platforms
While all NGS platforms perform sequencing of millions of small fragments of DNA or cDNA, there are several different sequencing technologies. Some platforms can produce more reads or different read lengths than others. The most prevalent and successful sequencing technology was pioneered by Illumina. Illumina’s instrument portfolio includes benchtop (iSeq, MiniSeq, MiSeq, NextSeq) and production-scale (NextSeq, HiSeq, and NovaSeq) sequencing platforms.
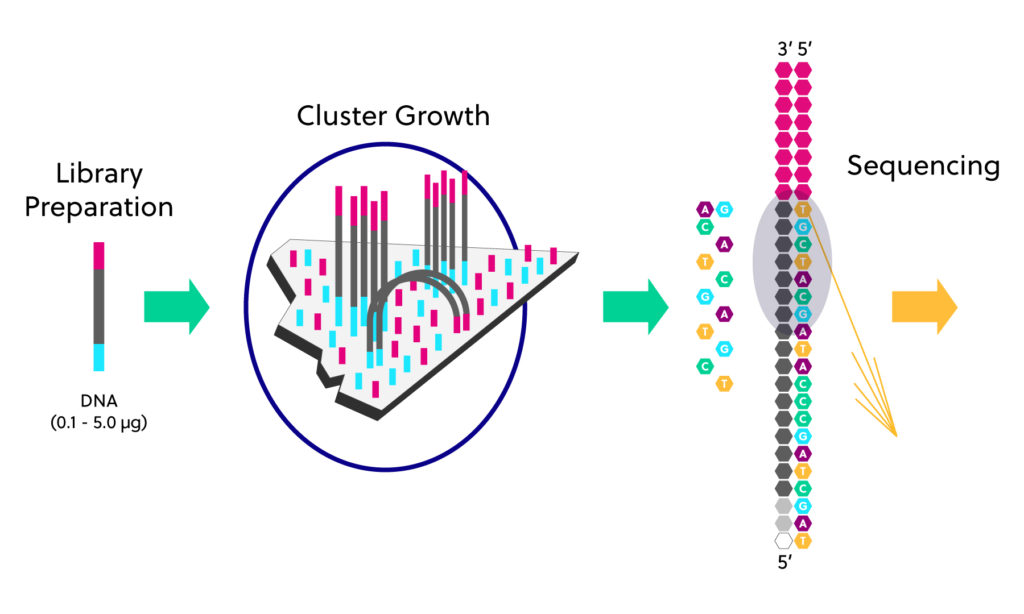
Illumina sequencers use a glass flowcell coated with millions of oligonucleotides that are complimentary to the sequencing adaptors.
Each library fragment hybridizes with the primers and is further amplified to generate millions to billions of clonal clusters. Then, fluorescently labeled nucleotides are used to synthesize a complementary strand for each fragment. After the addition of each tagged nucleotide, the flow cell is imaged and the emission from each cluster is recorded. The fluorescent emission wavelength and intensity are used to identify the sequence of the templates.
Step 4. Alignment and Data analysis
After sequencing is complete, specialized software is used to make sense of the large body of data produced. First the reads must be filtered for quality, amplicon size, and agreement between paired ends (for more on read length and paired end reads, stay tuned for our forthcoming page on NGS considerations). The reads are then assembled and aligned to a reference genome. Finally, reads (assembled or raw) can be compared to a reference sequence or to reads from another sample to identify variants based on disease state, etc. If reads are aligned with a reference genome, variant annotation can be used to associate variants with known genes or regulatory sequences. For information about bioinformatic analysis specifically related to the immune repertoire, see the Learning Center article describing iRepertoire’s data analysis platforms/services.
Sequencing in diagnostic/clinical applications
The application of sequencing to diagnostic and clinical applications has become more common as sequencing costs decrease. The data generated by NGS can be used to elucidate the mechanism that causes a disease, for clinical diagnosis, for therapeutic choice, and for prognosis. Compared to other methods of analysis such as protein staining-based methods, sequencing is more informative as it generates an array of details about the genome.
A large focus area for NGS is cancer treatment as the genetic composition of a particular tumor can help oncologists predict which treatments are likely to be effective. NGS can be used to identify tumor-specific variants such as copy number alterations, mutations, and gene expression alterations. Sequencing panels, which include primers that directly target particular transcripts or genomic regions of interest, are often used in oncology applications to reduce costs and more efficiently initiate genotype-directed targeted therapies. Based on sequencing results, patients may then undergo targeted therapy or immunotherapy.
As we continue to recognize the role the immune system plays in cancer response and the cancer micro-environment, immune repertoire sequencing is increasingly relevant for cancer studies. To learn more about immune repertoire sequencing, visit our pages which are specifically devoted to the topic.
Sequencing will no-doubt become more pervasive as new technologies are developed that automate the labor-intensive processes of library preparation and data analysis.