There are several factors to consider when planning a next generation sequencing (NGS) experiment. These considerations include: the depth of the sequencing coverage, the length of the sequencing reads, whether to conduct single-end versus paired-end sequencing, and multiplexing options. Because the cost of sequencing will vary based on these decisions, it’s important to plan your experiment for the appropriate amount of sequencing data required to answer your experimental questions. This post will help you navigate these considerations, particularly with regards to immune repertoire sequencing projects. For additional background information, please see our intro to NGS post.
Sequencing Coverage
Sequence coverage (depth) describes the average number of reads that align to a known reference at a particular location within the target transcript or genome. Because sequencing is error prone, higher coverage is used to increase confidence in the bases called. This approach is employed under the assumption that sequencing errors are random. Therefore, if each nucleotide is sequenced multiple times, the base call shared by the majority of reads (the consensus) will reflect the correct nucleotide.
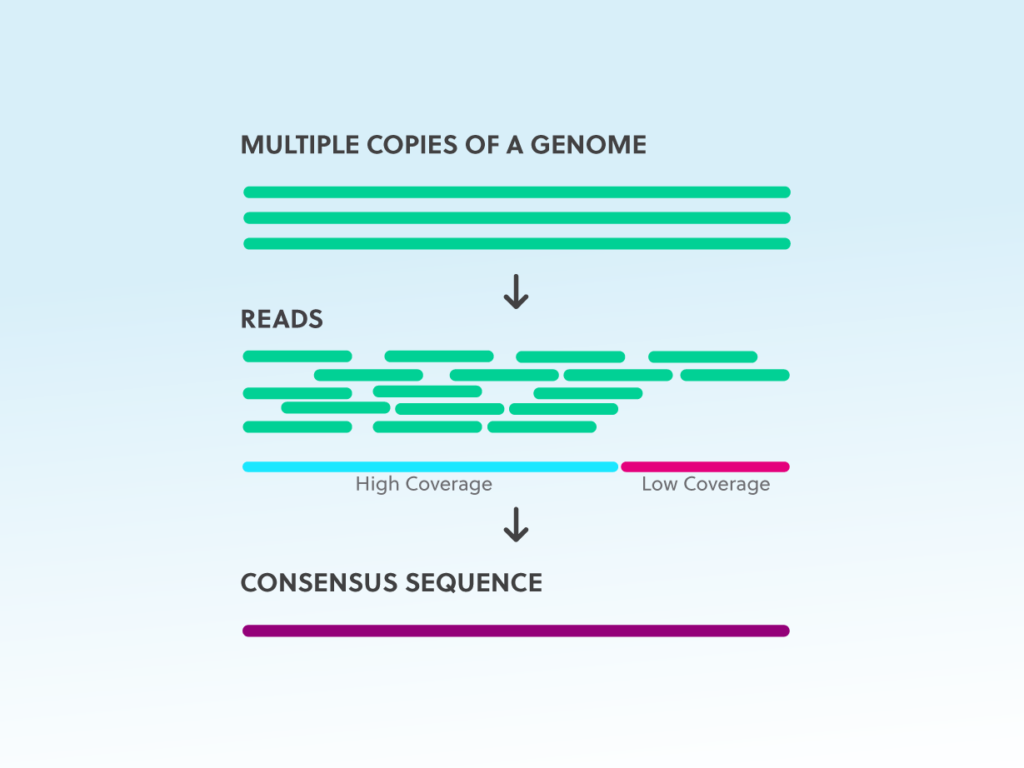
In NGS, sequence reads need to cover each base many times to increase the confidence; individual sequencing read errors are statistically irrelevant when they are outnumbered by correct reads. Coverage is variable within a sample and typical coverage ranges from 30 or less to >1000 reads for typical human genetic and cancer applications, respectively.
It is necessary to determine the sequencing coverage needed for your application to minimize the probability of false results. For instance, if you want to capture rare RNA sequences within the transcriptome, deeper sequencing will be needed to detect low abundance variants.
Immune repertoire sequencing is distinct from other sequencing applications because each cell has the potential to contain its own unique VDJ sequence. Thus, each cell represents a de novo rearrangement. During a targeted VDJ sequencing application, the VDJ rearrangement, and in particular the CDR3 region, is captured and sequenced directly so that no additional bioinformatic assembly of short read fragments is required (beyond potentially read stitching, discussed in the “Sequencing Read Length” section below). In an immune repertoire sequencing application, the sequencing coverage is determined based on the starting cell numbers of the sample and an estimate of sample diversity, if known. Sufficient reads need to be allotted for each sample to cover the potential diversity based on the input cell counts.
The average human white blood cell count ranges from 4,000 to 11,000 cells/µL. Cells are lost during all processing steps along the way, so it is important to have an estimation of the final cell count of the sample prior to RNA extraction. We generally recommend allocating a minimum of 5-10x the number of reads per the number of cells in the sample. Therefore, for a sample containing 100,000 cells, a minimum of 500,000 reads should be allocated. This means for samples with generally lower cell counts, there is an opportunity to pool more samples per lane (or flow cell), with the limit being the number of available molecular ids or barcodes for the chain of interest. If the sample, or the RNA source, is expected to have a restricted repertoire, it may be unnecessary to sequence very deeply. For instance, a Jurkat cell line has only one TCR-beta rearrangement. In this case, the cell count is irrelevant when determining the number of reads needed because the sequence can be obtained from very few reads. Assigning too many reads to such a sample wastes expensive flow cell real estate unnecessarily. Over-sequencing can also lead to a build-up of low frequency sequencing errors.
Sequencing Read Length
Read length describes the average length of the sequencing reads produced (i.e., the number of base pairs sequenced) and is sequencing-platform specific. If assembling the reads into the reconstructed DNA sequence is like doing a puzzle, long reads equate to larger puzzle pieces. For whole genome sequencing or species identification, longer reads are preferable. Longer read lengths are also essential for capturing insertions and deletions or for sequencing regions with a lot of redundancy, such as those that contain transposons.
Short reads are effective for applications aimed at counting the abundance of specific sequences, identifying variants within otherwise well-conserved sequences, or for profiling the expression of particular transcripts.
NGS read length and coverage are two parameters that can be controlled and need to be predetermined before sequencing. Since sequencing costs are incurred on a per-base-pair basis, there is typically a trade-off between sequencing depth and sequencing length. Sequencing platforms that perform longer reads will typically provide less coverage, a higher error rate, and higher cost per base relative to short read sequencing.
Sequencing read types: Single-end versus paired-end reading
The term “paired-end” read refers to the reading of both the forward and reverse template strands of the same DNA sequence during sequencing.
Because the distance between the two ends is known, this information can be used to map the reads over repetitive regions. Paired-end reading improves the ability to detect the relative position of sequencing reads and identify gene insertions, deletions, repetitive sequences, and other rearrangements. As it relates to the immune repertoire, paired-end reads can also be used to extend the coverage of the VDJ-receptor. Forward and reverse reads can be sequenced from both the V- and C- directions, which can be stitched together using a small area of overlap between the reads. This overlapping region can be used to extend the read length such that the receptor information can be provided from within framework 1 to the beginning of the C-region (for more information, see our post on iRepertoire’s primer systems and the technical notes for our bioinformatics platform).
Paired-end sequencing involves twice as much sequencing, so it’s more expensive, but it has the advantage of increased accuracy and extending the read length. As a result, the entire VDJ region can be captured directly without using any complex downstream genomic assembly. Single-end reads, which provide more coverage at the same cost, are ideal for studies where extending the read length is not important or increased accuracy can be provided through other means (for instance, unique molecular indexing). For VDJ analysis, data are lost during the process of read stitching; however, paired-end read data has the benefit of being analyzed as either single-end or paired-end. If the downstream application requires only the frequencies of the unique CDR3 region, then single-end data are sufficient for frequency calculations and CDR3 identification without suffering from data loss. However, if information needs to extended so that CDR1 and CDR2 regions are identified, the same data can be analyzed as paired-end with read stitching.
Multiplexing Libraries
A single sequencing run often produces more data than is necessary for a given project. To save costs, multiple samples can be combined into a sequencing run via pooling or multiplexing. In order to re-extract the individual samples from a pooled sequencing run, molecular barcodes are attached via the primers that are used to amplify the sequence during library preparation. Barcoding enables dozens or even hundreds of different samples to be multiplexed into a single sequencing run. Multiplexing is ideal when targeting a specific region of the genome in multiple different samples.
Data analysis software helps to demultiplex the sequencing data after the run by automatically sorting reads with different barcodes. Multiplexing samples into a single run means you will get less data for each sample. The coverage per sample will be roughly equal to the total coverage divided by the number of samples. For more information, see our post on library pooling.
Library preparation challenges
To prepare DNA for NGS, sequencing adapters may be ligated or added through PCR to the end of the DNA fragment. Library preparation workflows can include several rounds of amplification by PCR. Amplifying a mixture of templates is challenging because of the exponential nature of PCR. Non-specific or side product formation during the amplification can reduce the desired signal and contribute undesirable noise to the resultant data. For instance, small fragments are preferentially amplified; that means small amplicons can quickly take over the library, while large amplicons get excluded. Excluding (through experimental design) or removing (through purification steps) unwanted material during library preparation reduces noises from the PCR amplification; this can include unwanted or artefactual small amplicons such as primer-dimers. This results in increased signal from the desired product, and ultimately, better data.
To learn about how iRepertoire’s sequencing technology excludes unwanted amplicons, including primer-dimers, see our amplification technology post.